国際共同第Ⅲ相継続投与試験(ARGX-113-1803/ADVANCE+試験)※
(ARGX-113-1801の継続投与試験)1-4)
※本試験は実施中であり、1回目の中間解析(中間解析1:試験の概要、安全性及び有効性は2022年9月28日、薬物動態、薬力学及び免疫原性は2022年8月10日をデータカットオフ日とした解析)に基づいて記載。
「禁忌を含む注意事項等情報」等は電子添文をご参照ください。
国際共同第Ⅲ相継続投与試験(ARGX-113-1803/ADVANCE+試験)※
(ARGX-113-1801の継続投与試験)1-4)
※本試験は実施中であり、1回目の中間解析(中間解析1:試験の概要、安全性及び有効性は2022年9月28日、薬物動態、薬力学及び免疫原性は2022年8月10日をデータカットオフ日とした解析)に基づいて記載。
成人持続性及び慢性ITP患者を対象にウィフガート®の長期静脈内投与時の安全性及び有効性を評価する。
国際共同第Ⅲ相試験(ARGX-113-1801試験)の継続投与試験、長期、単群、非盲検、多施設共同(欧州、米国、ロシア、トルコ、ウクライナ、日本)
ARGX-113-1801試験に参加し、継続投与試験に移行した成人ITP患者101例(日本人5例)
ウィフガート®群から63例(日本人3例)、プラセボ群から38例(日本人2例)
ARGX-113-1801試験で24週間の治験薬投与期を完了した成人ITP患者は、本試験に移行可能とした。

ウィフガート®10㎎/㎏を週1回又は2週に1回、1時間かけて静脈内投与した。ウィフガート®10㎎/㎏の投与頻度は、ARGX-113-1801試験での投与頻度(週1回又は2週に1回)を継続した。血小板数に基づく投与頻度の変更はベースラインから許容されていたため、患者は52週間の治験薬投与期中のいつでも、週1回又は2週に1回のいずれかの頻度で投与を受けることが可能であった※3。試験開始時に投与されていた併用ITP治療薬※1及びレスキュー治療※4が許容された。
最初の52週間の治験薬投与期を完了した患者は、さらに最大3回の52週間の治験薬投与期、合計で最大4年間の治験薬投与(及び8週間のフォローアップ期)への参加を可能とした。
※1:併用ITP治療薬(経口副腎皮質ステロイド†、経口免疫抑制剤†、ダナゾール‡、ジアフェニルスルホン‡、ホスタマチニブ、経口TPO-RA†)は、ホスタマチニブ及び経口TPO-RAの承認用法・用量に基づく用量調整等が許容されたことを除き、追加及び増量は禁止された。血小板数が100,000/μLを超えた場合にのみ、医師の判断で減量又は中止が可能とされた。
†:ITP治療に対し本邦未承認の薬剤を含む、‡:ITP治療に対し本邦未承認
※2:8~11週の血小板数の測定で4回とも血小板数が30,000/μL未満の場合は効果不十分として早期中止した。
※3:連続した4回の来院(4回目の来院が判定時の来院)のうち3回で血小板数が100,000/μL以上となり、かつこれら4回の最後の来院時に100,000/μL以上になった場合、又は血小板数が連続した3回の来院で100,000/μL以上になった場合に投与頻度を2週に1回にした。
2週に1回の投与中、連続した2回の来院で血小板数が100,000/μL未満又は1回の来院で血小板数が30,000/μL未満になった場合、又はレスキュー治療を受けた患者は、投与頻度を2週に1回から週1回に増やした。
血小板数が400,000/μLを超えた場合は投与を一時中断し、血小板数が150,000/μL未満に減少したことを確認した上で2週に1回の投与頻度で再開した。
※4:血小板数が30,000/μL未満で、以下のいずれかに該当する患者に対しては、レスキュー治療が許可された。
・差し迫った出血リスク又は臨床的に重大な出血又は粘膜出血
・緊急手術の必要性
有害事象、特に注目すべき有害事象(AESI)※1、重篤な有害事象、バイタルサイン、臨床検査値などの評価
最初の52週間の治験薬投与期中に収集されたデータについて有効性解析を実施した。
※1:19~24週の6回の来院のうち4回以上で血小板数50,000/μL以上を達成
※2:17~24週の8回の来院のうち6回以上で血小板数50,000/μL以上を達成
主要評価項目及び副次評価項目は、ARGX-113-1801試験での投与群別(ウィフガート®群及びプラセボ群)、並びに試験全体として記述的に要約した。
すべてのカテゴリーの評価項目(有害事象、血小板数反応)及び追加解析である6週間間隔で複数の閾値を用いた持続的血小板数反応について、頻度表を作成した。
特に記載のない限り、有害事象及び臨床検査値異常はいずれも治験薬による治療下で発現したものとした。
中間解析のデータカットオフは1回目を2022年9月28日とした。
本試験では、先行試験でウィフガート®を投与された患者集団をウィフガート®-ウィフガート®群と表示し、先行試験でプラセボを投与された患者集団をプラセボ-ウィフガート®群と表示しました。両群を合わせて、全体集団と表示しました。
2022年9月28日データカットオフ時点でARGX-113-1801試験での治験薬投与及び試験を完了した106例中101例が本試験に移行し、すべての患者がウィフガート®の投与を1回以上受けました。このうちウィフガート®-ウィフガート®群が63例、プラセボ-ウィフガート®群が38例でした。
| 全体集団(n=101) | |
|---|---|
| 年齢(歳)、中央値(範囲) | 50.0(19-87) |
| 18歳以上65歳未満、例数(%) | 81(80.2%) |
| 性別、例数(%) | |
| 女性 | 49(48.5%) |
| 男性 | 52(51.5%) |
| 人種、例数(%) | |
| 白人 | 96(95.0%) |
| ITPの分類、例数(%) | |
| 慢性ITP(診断から12カ月超) | 89(88.1%) |
| 最初の診断からの期間(年)、中央値(範囲) | 4.0(0.3-54.1) |
| ARGX-113-1801試験のベースラインからの血小板数/μL、中央値(範囲) | 17,000(0-51,000) |
| ウィフガート®-ウィフガート®群(n=63) | 17,000(0-51,000) |
| プラセボ-ウィフガート®(n=38) | 12,500(2,000-31,000) |
| ITP前治療の数 3種類以上、例数(%) | 71(70.3%) |
| 出血性イベント(WHOスケール)Grade 1以上、例数(%) | 53(52.5%) |
治験薬への曝露(2022年9月28日データカットオフ時点)
ウィフガート®の投与回数の中央値(範囲)は23.0(3-71)回で、31例(30.7%)は2週に1回の投与頻度に1回以上変更しました。
有害事象(全体集団)
全体集団における有害事象は101例中93例(92.1%)に認められました。
主な有害事象(全体集団の10%以上)
尿中血陽性42例(41.6%)、COVID-19 20例(19.8%)、点状出血17例(16.8%)
重篤な有害事象
12例(11.9%)21件に認められました。嘔吐2例(2.0%)2件、慢性腎臓病1例(1.0%)2件、心筋梗塞、下痢、胃炎、悪心、直腸出血、全身性炎症反応症候群、COVID-19、COVID-19肺炎、大腿骨骨折、ヘモグロビン減少、血小板数減少、基底細胞癌、肺の悪性新生物、脳出血、頭痛、急性呼吸不全、肺線維症各1例(1.0%)1件でした。
投与中止に至った有害事象
慢性腎臓病1例(1.0%)1件でした。
死亡に至った有害事象
脳出血、肺線維症、大腿骨骨折が各1例に認められ、いずれも治験薬との因果関係は関連なしと判断されました。
AESIと定義した出血性事象
76例(75.2%)293件に報告されました。5%以上報告されたAESIは、尿中血陽性、点状出血、鼻出血、挫傷、血腫、歯肉出血、斑状出血、口腔内出血、血尿、紫斑でした。CTCAE Grade 3以上のAESIは2例(2.4%)2件に報告され、直腸出血、脳出血が各1件でした。これらのAESIはいずれも重篤であり、脳出血は死亡に至りました。
AESIと定義した感染症
34例(33.7%)53件に報告されました。5%以上報告されたAESIは、COVID-19でした。CTCAE Grade 3以上のAESIはCOVID-19(Grade2)及びCOVID-19肺炎を発現した1例であり、重篤でした。
臨床検査値(全体集団)4)
多く報告されたCTCAE Grade 3以上の異常値は高カリウム血症、リンパ球数減少、好中球数減少、白血球数減少であり、各3例(3.0%)でした。Grade 4の異常値は、高カリウム血症3例、腎クレアチニン・クリアランス減少、クレアチニン増加、リンパ球数減少、好中球数減少の各1例でした。
| 事象 | ウィフガート®-ウィフガート®群 (n=63) | プラセボ群-ウィフガート®群 (n=38) | 全体集団 (n=101) |
|---|---|---|---|
| 例数(%) | 例数(%) | 例数(%) | |
| 発現例数(発現率) | 7(11.1) | 4(10.5) | 11(10.9) |
| 血液およびリンパ系障害 | 2(3.2) | 1(2.6) | 3(3.0) |
| 貧血 | 1(1.6) | 0 | 1(1.0) |
| 好酸球増加症 | 0 | 1(2.6) | 1(1.0) |
| 白血球減少症 | 1(1.6) | 0 | 1(1.0) |
| リンパ球増加症 | 0 | 1(2.6) | 1(1.0) |
| 胃腸障害 | 1(1.6) | 0 | 1(1.0) |
| 腹痛 | 1(1.6) | 0 | 1(1.0) |
| 一般・全身障害および投与部位の状態 | 0 | 1(2.6) | 1(1.0) |
| 注射部位異常感覚 | 0 | 1(2.6) | 1(1.0) |
| 臨床検査 | 1(1.6) | 0 | 1(1.0) |
| 血中ビリルビン増加 | 1(1.6) | 0 | 1(1.0) |
| 筋骨格系および結合組織障害 | 1(1.6) | 1(2.6) | 2(2.0) |
| 背部痛 | 0 | 1(2.6) | 1(1.0) |
| 筋痙縮 | 1(1.6) | 0 | 1(1.0) |
| 神経系障害 | 2(3.2) | 1(2.6) | 3(3.0) |
| 頭痛 | 2(3.2) | 1(2.6) | 3(3.0) |
| 皮膚および皮下組織障害 | 1(1.6) | 0 | 1(1.0) |
| 斑状丘疹状皮疹 | 1(1.6) | 0 | 1(1.0) |
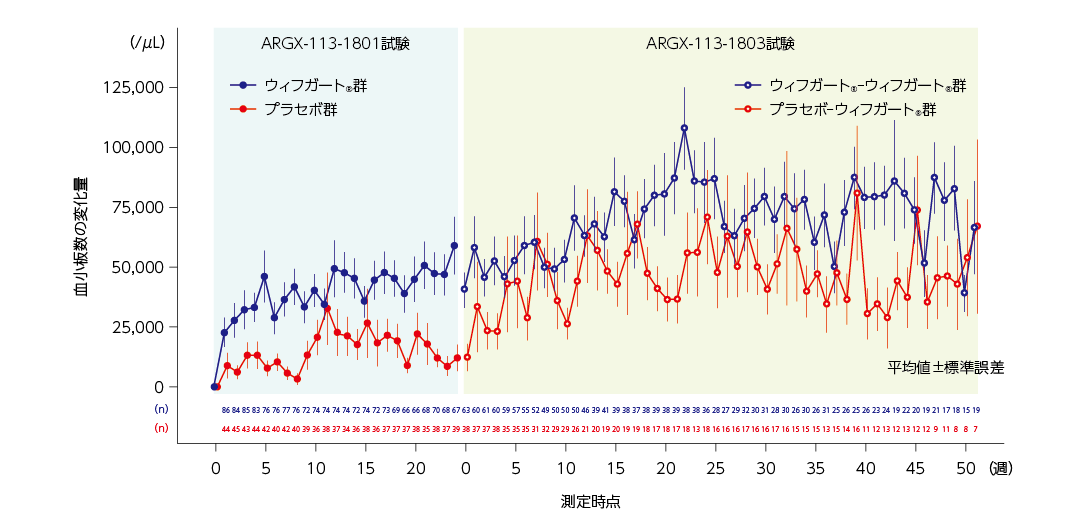
各来院時の血小板数のベースラインからの平均変化量(2022年9月28日データカットオフ時点)
ウィフガート®-ウィフガート®群では、血小板数のARGX-113-1801試験のベースラインからの平均変化量(標準誤差)は、最初の52週間の治験薬投与期を通して38,830(7,761)/μLから107,690(17,025)/μLの間で維持されました。
プラセボ-ウィフガート®群では、平均血小板数は1週の時点でARGX-113-1801試験のベースラインから増加し、平均変化量(標準誤差)は33,310(18,579)/μLでした。2週以降、平均血小板数(標準誤差)は、最初の52週間の治験薬投与期を通して23,060(7,301)/μLから80,640(27,939)/μLの間で維持されました。
ウィフガート®を初めて投与された患者における持続的血小板数反応が認められた患者の割合(2022年9月28日データカットオフ時点)
プラセボ-ウィフガート®群38例において、19~24週の6回の来院のうち4回以上で血小板数50,000/μL以上を達成した患者は10例(26.3%)、17~24週の8回の来院のうち6回以上で血小板数50,000/μL以上を達成した患者は8例(21.1%)でした。
〈追加評価〉(2022年9月28日データカットオフ時点)
規定した6週間間隔(1~6週、7~12週など、43~48週まで)で、6回の来院のうち4回以上で血小板数50,000/μL以上を達成し、持続的血小板数反応が認められた患者を評価しました。
1)社内資料:第3相試験ARGX-113-1803試験(承認時評価資料)(CTD2.7.6.4)(EFG90104)
2)社内資料:1803試験(第3相、1801試験の非盲検継続投与試験)中間解析1(承認時評価資料)(CTD2.7.3.2.2)(EFG90096)
3)社内資料:ベースラインからの血小板数の変化(承認時評価資料)(CTD2.7.3.3.2.1.4)(EFG90098)
4)社内資料:1801試験及び1803試験(承認時評価資料)(CTD2.7.4.3.2)(EFG90100)
JP-VJITP-25-00537(2025年11月作成)
