目的
成人CIDP患者に対するヒフデュラ®の有効性、安全性及び忍容性を評価する
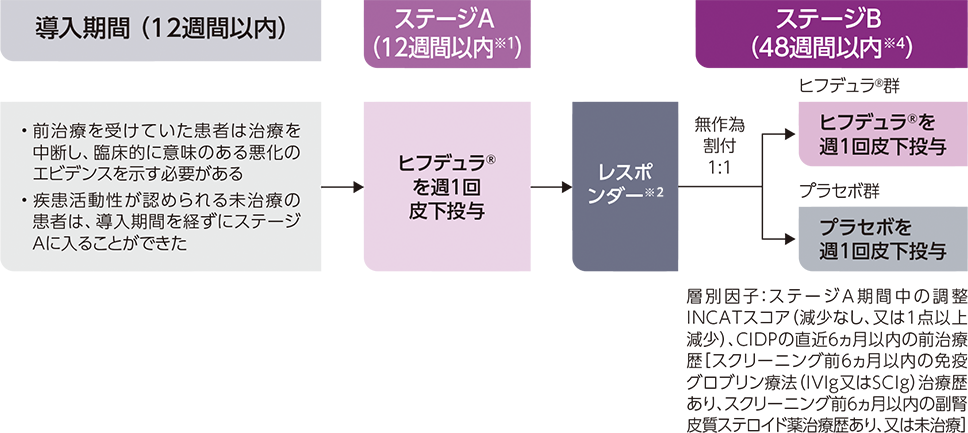
試験デザイン
第Ⅱ相、多段階、多施設共同(日本を含む22か国の146施設)
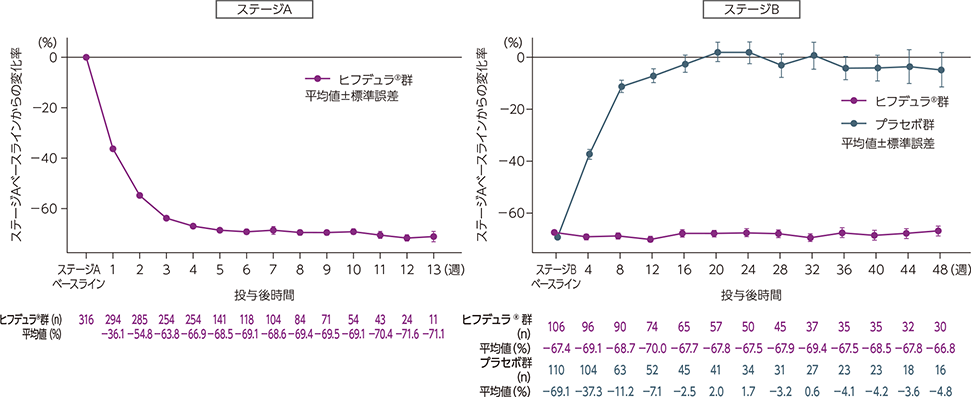
ステージA:非盲検
ステージB:ランダム化治療中止、二重盲検、プラセボ対照
対象
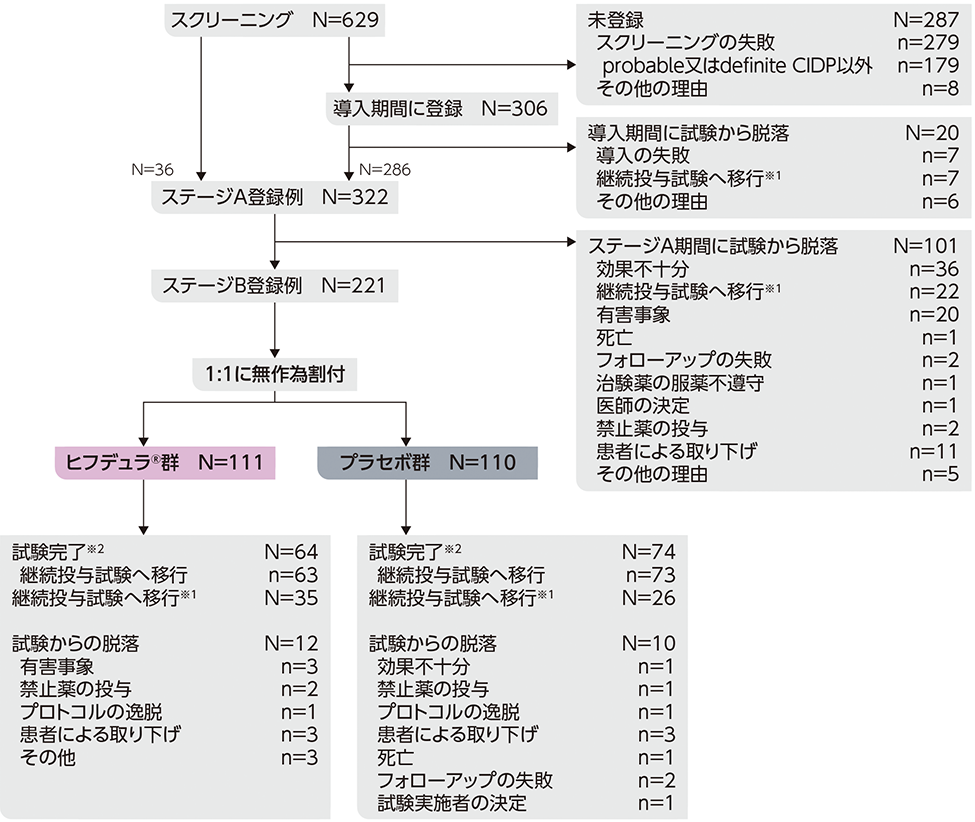
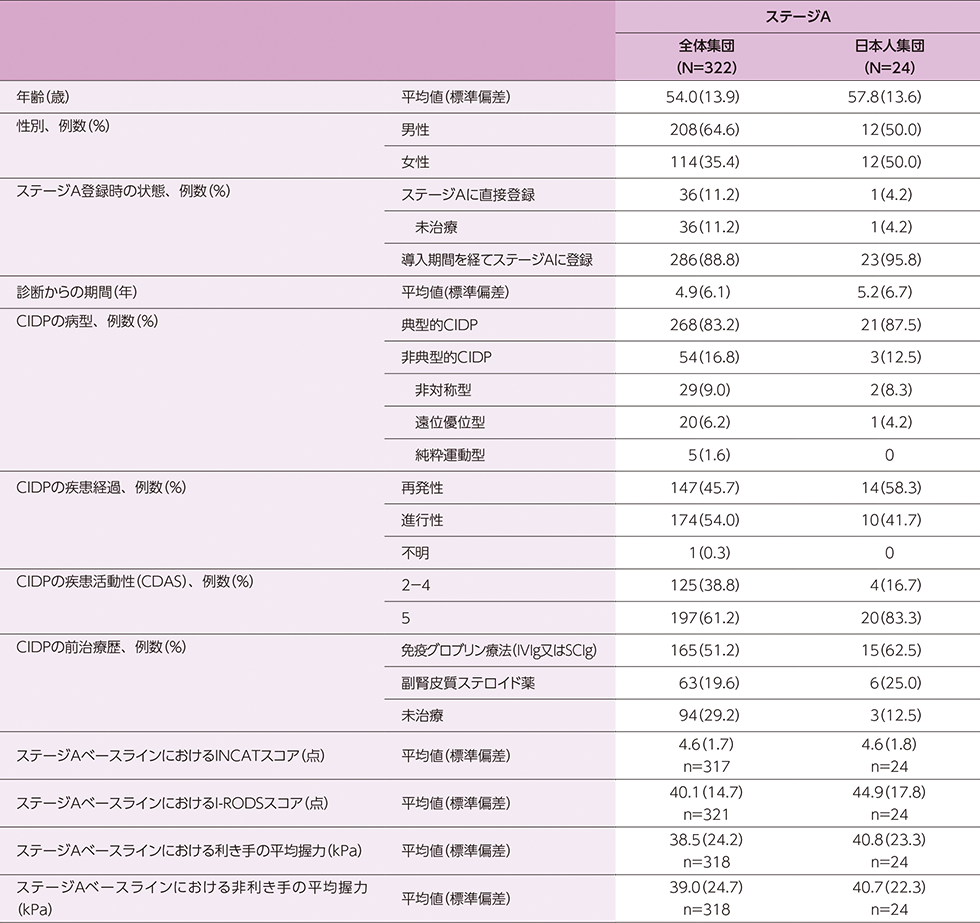
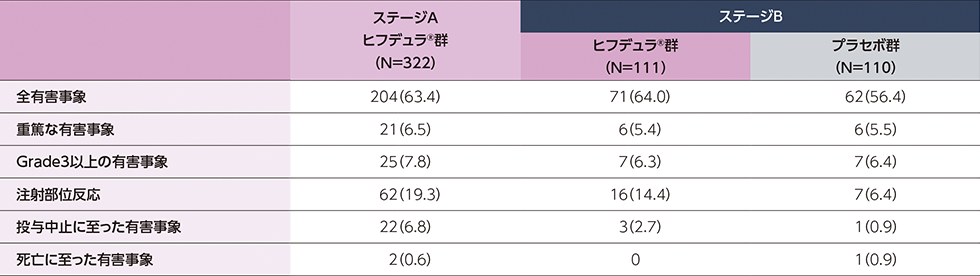
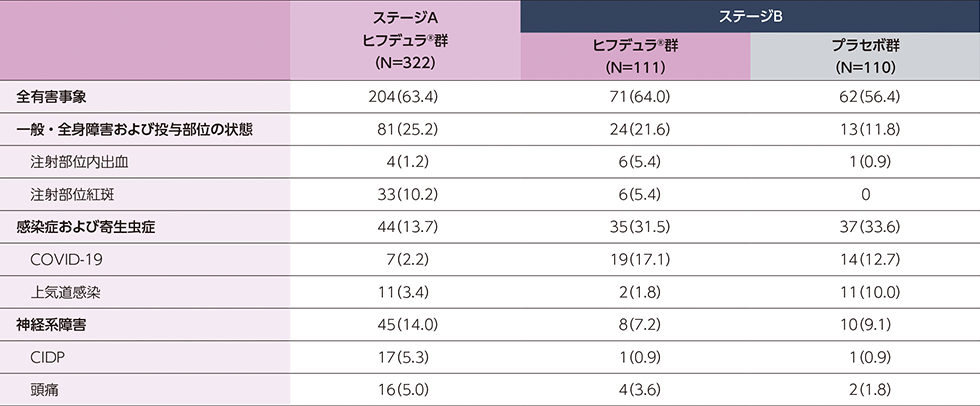
probable又はdefinite CIDPと診断された進行性又は再発性のCIDP患者322例(日本人24例)
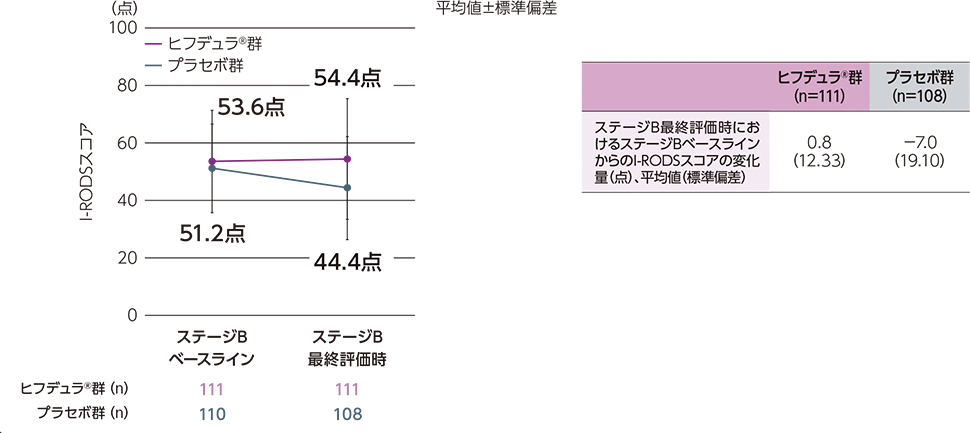
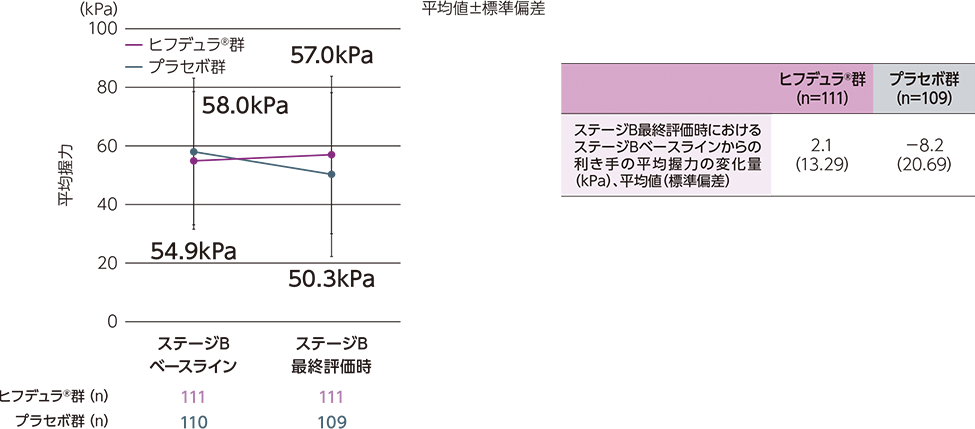
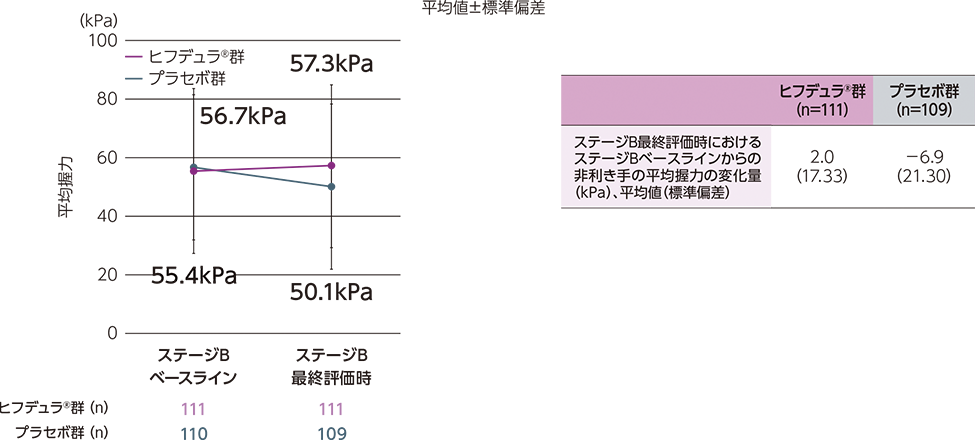
ステージAのレスポンダー※はステージBに移行し、ヒフデュラ®群とプラセボ群に1:1で無作為割付された
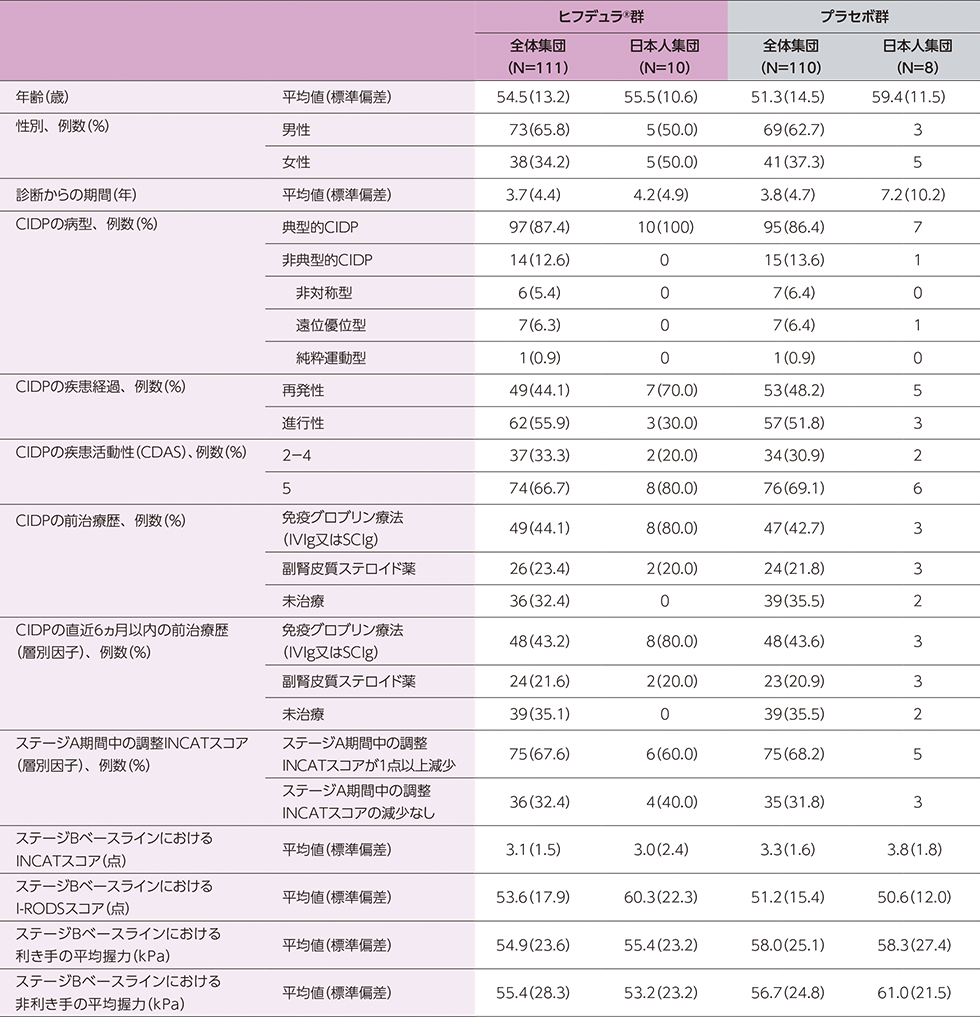
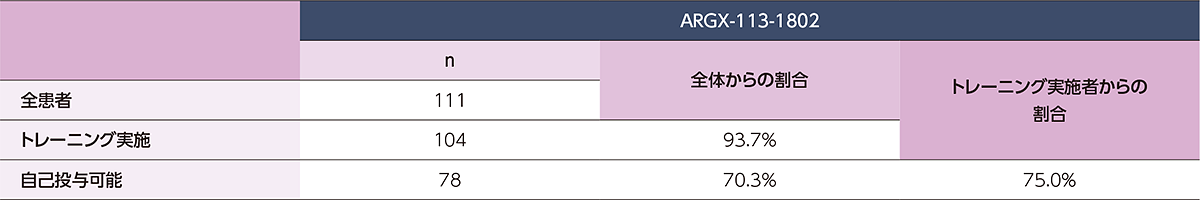
(ステージA:322例、ステージB ヒフデュラ®群:111例、ステージB プラセボ群:110例)
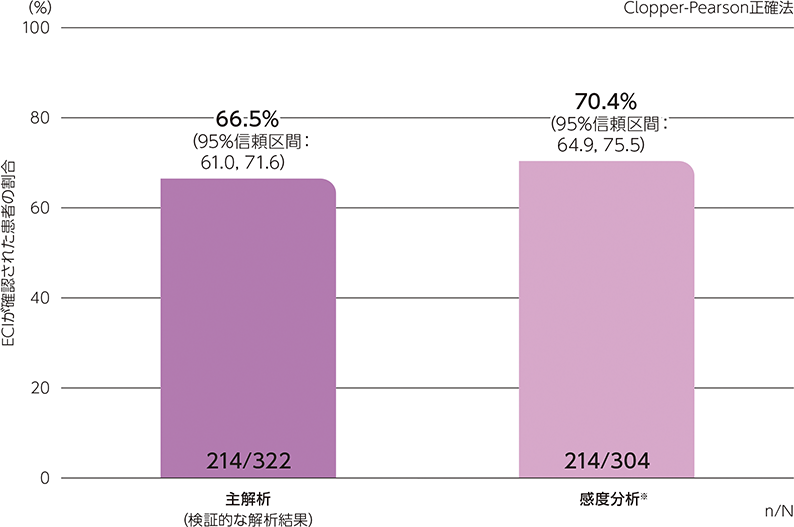
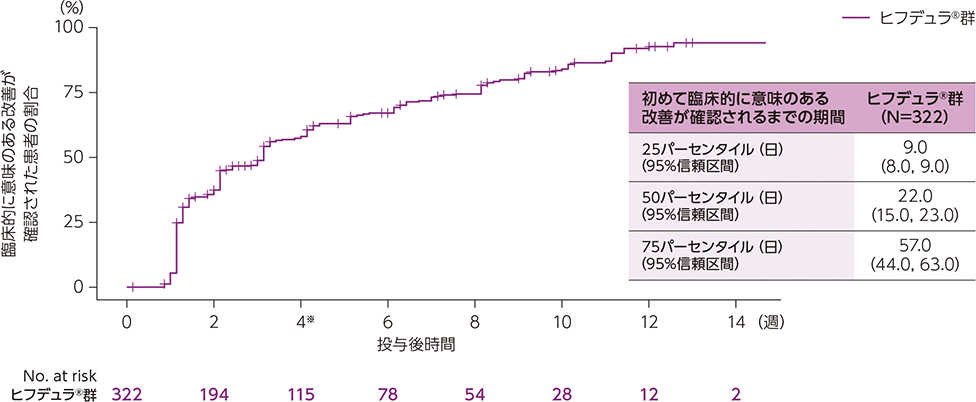
※ ステージAで臨床的改善のエビデンス(ECI)が確認された患者
主な選択基準
- 18歳以上で、European Federation of Neurological Societies/Peripheral Nerve Society(EFNS/PNS)の診断基準(2010)に基づいてprobable又はdefinite CIDPと診断され、進行性又は再発性の患者
- スクリーニング時点で、CIDP Disease Activity Status(CDAS)スコアが2点以上であった患者
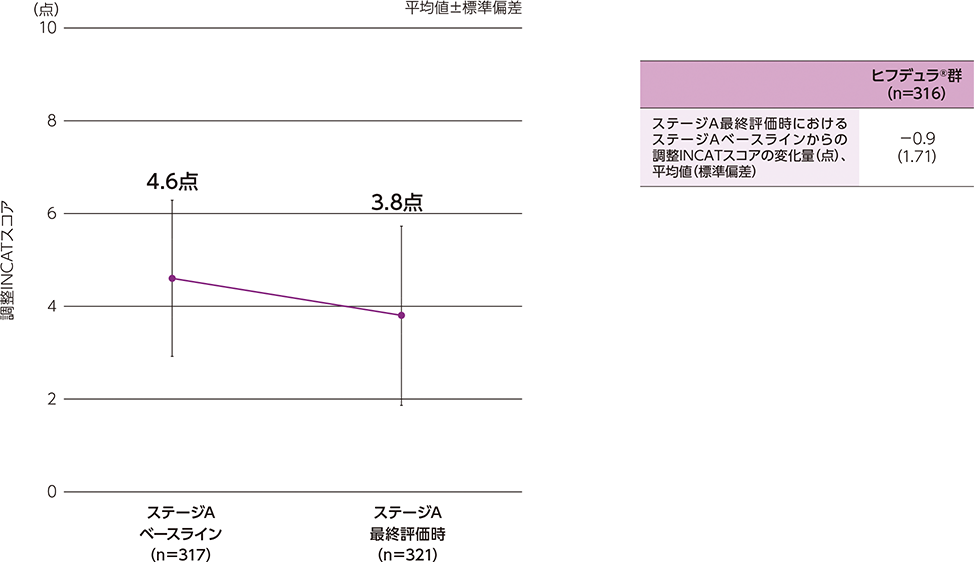
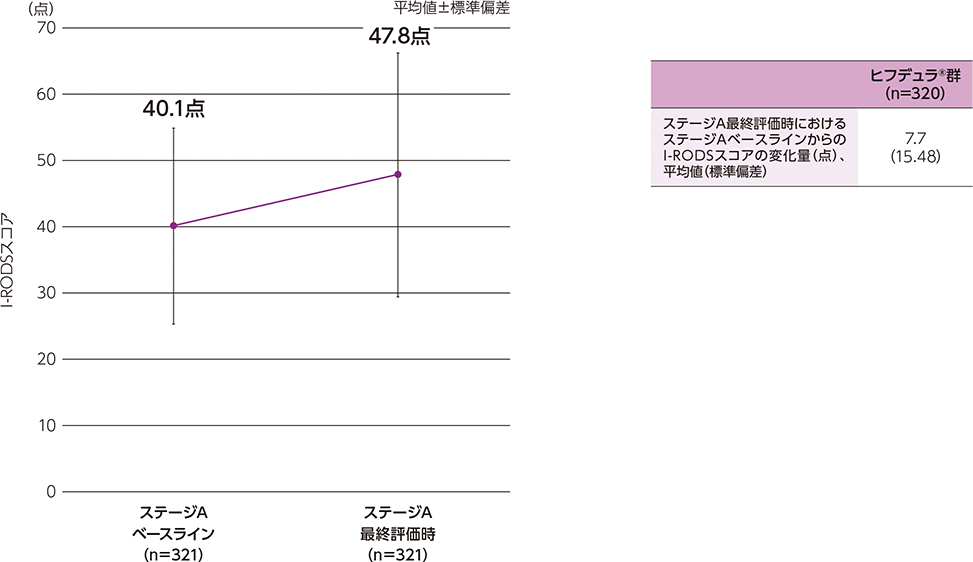
- 導入期間の初回来院時又はステージAベースラインで、INCATスコアが2点以上であった患者
- 以下のいずれかの治療状況を満たしている場合
- スクリーニング前6ヵ月以内に、ステロイドパルス療法、プレドニゾロン/prednisone注1) 10mg/日換算量以下の経口副腎皮質ステロイド薬、又は免疫グロブリン療法(IVIg又はSCIg)注2)の治療を受けており、導入期間の初回来院時にこの治療法を中止する意思がある
- 未治療(過去にCIDPの治療歴がないか、副腎皮質ステロイド薬、免疫グロブリン療法(IVIg又はSCIg)注2)による治療をスクリーニング前6ヵ月以上受けていない)
注1)prednisoneは本邦未承認である。
注2)一部の副腎皮質ステロイド薬、IVIg及びSCIgはCIDPに対して本邦未承認である。
投与方法
- ステージA:ヒフデュラ®注)を週1回皮下投与した*(12週間以内※1)
- ステージB:ステージAのレスポンダー※2をヒフデュラ®注)群とプラセボ群※3に1:1で無作為割付し、週1回皮下投与した*(48週間以内※4)
*ヒフデュラ®はバイアル製剤を使用した。